Ein Überblick über das Dravet-Syndrom

Die Krankheit ist mit einem genetischen Defekt im SCN1A-Gen verbunden, obwohl sie ohne den genetischen Defekt auftreten kann. Die Diagnose basiert auf den klinischen Anzeichen und Symptomen eines Kindes. Die Diagnose kann durch diagnostische Tests unterstützt werden.
Symptome
Krampfanfälle sind das früheste Symptom des Dravet-Syndroms. Entwicklungsprobleme sowie Krampfanfälle verschlimmern sich im Allgemeinen, wenn ein Kind älter wird.Die Symptome des Dravet-Syndroms umfassen:
- Anfälle: Krampfanfälle sind oft mit Fieber verbunden, obwohl sie ohne Fieber auftreten können. Es gibt verschiedene Anfallstypen, die typischerweise beim Dravet-Syndrom auftreten, einschließlich myoklonischer Anfälle, tonischer klonischer Anfälle und nicht konvulsiver Anfälle. Längere Anfälle und Status epilepticus sind ebenfalls charakteristisch für die Erkrankung. Tatsächlich kann der erste Anfall eine besonders lange Dauer haben.
- Anfallsauslöser: Menschen mit Dravet-Syndrom leiden möglicherweise unter Krampfanfällen, bei denen es zu Krampfanfällen infolge heller oder blinkender Lichter kommt. Darüber hinaus kann eine Person mit Dravet-Syndrom anfällig für Anfälle sein, wenn sich die Körpertemperatur ändert.
- Ataxie (Gleichgewichtsstörungen): Schwierigkeiten bei der Koordination und beim Gehen, die als Ataxie bezeichnet werden, beginnen in der Kindheit und setzen sich in der Jugend und im Erwachsenenalter fort.
- Motorische Beeinträchtigung: Bei Menschen mit Dravet-Syndrom wird eine geduckte Position beim Gehen beschrieben. Oft ist ein niedriger Muskeltonus vorhanden, der sich als Muskelschwäche äußern kann.
- Kognitive Beeinträchtigung: Kinder können Sprach- und kognitive Probleme entwickeln, die ein Leben lang anhalten. Beim Dravet-Syndrom kann es eine Reihe kognitiver Fähigkeiten geben, und einige Betroffene haben eine höhere Lernfähigkeit als andere.
- Verhaltensprobleme: Kinder und Erwachsene, die mit dem Dravet-Syndrom leben, können eine Reizbarkeit, Aggression oder ein Verhalten aufweisen, das an Autismus erinnert.
- Infektionen: Menschen mit Dravet-Syndrom sind anfällig für Infektionen.
- Unregelmäßigkeiten bei der Schweiß- und Temperaturregulierung: Menschen mit Dravet-Syndrom können Veränderungen im autonomen Nervensystem erfahren, die insbesondere vermindertes Schwitzen und zu hohe oder zu niedrige Körpertemperaturen verursachen.
- Knochenprobleme: Das Dravet-Syndrom ist mit gebrechlichen Knochen und einer Veranlagung für Knochenbrüche verbunden.
- Herzrhythmusstörungen: Etwa ein Drittel der Menschen mit Dravet-Syndrom haben einen unregelmäßigen Herzschlag, wie eine schnelle Herzfrequenz, eine langsame Herzfrequenz oder eine andere Unregelmäßigkeit, wie ein verlängertes QT-Intervall.
Das Dravet-Syndrom ist eine lebenslange Erkrankung und die Symptome bessern sich nicht. Es besteht ein erhöhtes Risiko für einen frühen Tod, häufig im Zusammenhang mit Verletzungen aufgrund von Anfällen. Bei Menschen mit Dravet-Syndrom ist die Wahrscheinlichkeit eines plötzlichen unerwarteten Todes bei Epilepsie (SUDEP), einem Zustand, bei dem ein unerwarteter Tod auftritt, in der Regel im Schlaf, ebenfalls höher.
Ursachen
Es wird angenommen, dass das Dravet-Syndrom durch einen Defekt in der Funktion der Natriumkanäle verursacht wird und als eine Form der Kanalopathie beschrieben wird. Natriumkanäle regulieren die Gehirn- und Nervenfunktion. Ein Defekt in der Funktion von Natriumkanälen kann eine Vielzahl von Problemen verursachen, einschließlich einer unregelmäßigen Gehirnaktivität, die sich als Anfälle manifestiert, und einer fehlerhaften Kommunikation zwischen Gehirnzellen, die sich als Entwicklungsstörung manifestiert.Genetik
Ungefähr 80 Prozent der Menschen mit Dravet-Syndrom haben einen Chromosom-2-Defekt im SCN1A-Gen, das für Natriumkanäle kodiert. Dieser Defekt tritt in einem erblichen Muster auf, und die Mutation kann auch zum ersten Mal bei einem betroffenen Kind auftreten.
Diagnose
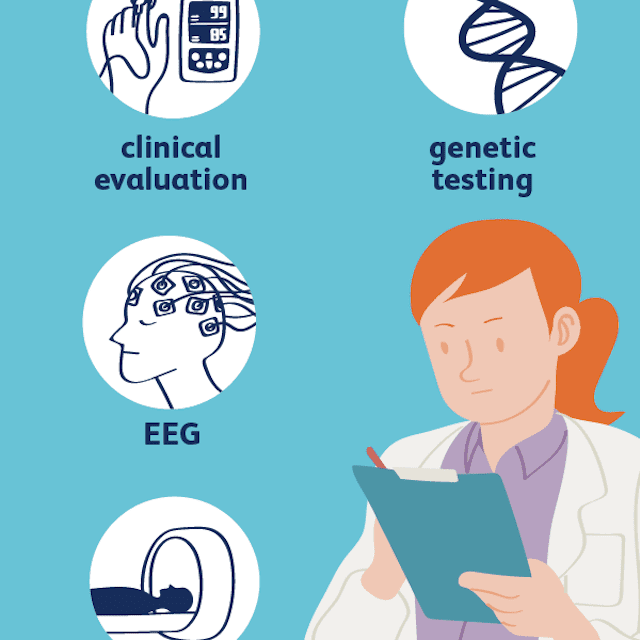
Das Dravet-Syndrom wird anhand der klinischen Beurteilung eines Arztes diagnostiziert. Diagnosestudien können die Diagnose stützen, aber nicht bestätigen oder ausschließen. Nach Angaben der Dravet-Syndrom-Stiftung umfassen die klinischen Merkmale des Dravet-Syndroms mindestens vier der folgenden fünf Merkmale:- Normale kognitive und motorische Entwicklung vor dem ersten Anfall
- Zwei oder mehr Anfälle vor dem ersten Lebensjahr
- Eine Kombination aus myoklonischen, hemiklonischen oder generalisierten tonisch-klonischen Anfällen
- Zwei oder mehr Anfälle, die länger als 10 Minuten dauern
- Mangelnde Besserung durch die Standardbehandlung mit Antikonvulsiva und anhaltende Anfälle nach dem zweiten Lebensjahr
- Elektroenzephalogramm (EEG): Das EEG ist in der Regel normal, wenn eine Person mit Dravet-Syndrom keinen Anfall hat, insbesondere bei sehr kleinen Kindern. Das EEG zeigt Anomalien im Zusammenhang mit der Anfallsaktivität während eines Anfalls. In der späteren Kindheit und während des gesamten Jugend- und Erwachsenenalters kann das EEG auch ein Muster der Verlangsamung zwischen Anfällen aufweisen.
- Gehirn-MRT: Typischerweise wird erwartet, dass die Hirn-MRT einer Person mit Dravet-Syndrom normal ist. Im Erwachsenenalter kann es zu einer Atrophie (Ausdünnung) des Hippocampus oder des gesamten Gehirns kommen.
- Gentest: Gentests können die SCN1A-Mutation identifizieren, die am häufigsten bei Menschen mit Dravet-Syndrom auftritt. Es wurde in einem Mosaikmuster vermerkt, was bedeutet, dass eine Person einige Zellen mit der Mutation haben kann, und einige ohne sie.
Behandlung
Es gibt eine Reihe von verschiedenen Problemen, die eine Person mit Dravet-Syndrom zu erwarten hat, und alle sind schwer zu behandeln. Die Behandlung der physischen, kognitiven und Verhaltensprobleme des Dravet-Syndroms ist individualisiert und kann Physiotherapie, Sprachtherapie und Verhaltenstherapie umfassen.Die Anfälle sind nicht leicht zu kontrollieren. Typischerweise umfassen Antikonvulsiva, die beim Dravet-Syndrom verwendet werden, eine Kombination aus Valproat, Clobazam, Stiripentol, Topiramat, Levetiracetam und Bromiden. Eine ketogene Diät und eine Vagusnervstimulation werden auch für die Behandlung der Anfälle in Betracht gezogen.
Cannabidiol
Im Juni 2018 genehmigte die US-amerikanische Food and Drug Administration (FDA) Epidiolex (Cannabidiol) zur Behandlung des Dravet-Syndroms sowie eines weiteren Epilepsiesyndroms, des Lennox-Gastaut-Syndroms. Frühere Studien zeigten, dass Kinder mit Dravet-Syndrom unter Epidiolex eine Abnahme der Anfallshäufigkeit erlebten und in der Lage waren, die Medikation zu tolerieren.
Medikamente, die das Dravet-Syndrom verschlimmern
Zu den üblichen Antikonvulsiva, von denen angenommen wird, dass sie die Natriumkanäle beeinflussen, gehören Carbamazepin, Oxcarbazepin, Phenytoin und Lamotrigin. Diese können Anfälle beim Dravet-Syndrom eher verschlimmern als verbessern.
Ein Wort von Verywell
Wenn bei Ihrem Kind das Dravet-Syndrom diagnostiziert wurde, ist dies eine schwierige Situation. Ihr Kind wird während seines gesamten Lebens ein enges Management und eine sorgfältige Betreuung benötigen. Viele der Symptome des Dravet-Syndroms können sich bei richtiger Behandlung teilweise bessern. Die Behandlungsstrategien müssen möglicherweise geändert werden, wenn Ihr Sohn oder Ihre Tochter physisch wächst und wenn sich ihr Zustand mit zunehmendem Alter verbessert oder verschlechtert.Wie bei vielen seltenen Krankheiten kann das Gefühl der Isolation und des Nichtwissens, was zu erwarten ist, überwältigend sein. Einige Familien finden es hilfreich, über Selbsthilfegruppen und Patientenvertretungsgruppen mit anderen in Kontakt zu treten, die möglicherweise die gleichen Probleme haben.